How to make big bio molecules and molecular units selectively visible ?
Starting point:
- some big molecule (f.i. protein, enzyme or receptor, hormone, DNA, RNA) is known to the researcher, its composition, or at least an important, typical part of it is understood.
- On the other side there's the material to be analyzed, a suspension of molecular material, maybe from a living or dead cell or tissue or maybe just one big soup consisting out of all these alternatives.
- the question arises: is there at least one type of molecules one is looking for available in the material analyzed? If the answer is yes, where is this molecule (or molecules, if one is looking for more than just one distinct type) localized?
- Problem to be solved: there's no microscope or any other scope, that would allow one to see clearly and thus also find this kind of molecules. The problem is not so much the spatial resolution (the raster tunneling microscopes one can look at single atoms) . It's the required selectivity: there's no direct way to find those molecules among millions and billions of similar molecules in the sample.
- A motivating help for scientists: In the human, animal, plant and in general cell genetics we would like to find out on which chromosome and where on that specific chromosome certain genes and gene groups are localized and how their addition to the genome has happened.
- A motivating help for diagnosticians: histo pathology and cell pathology expect extraordinary specificity and selective power from methods used to prove the presence of AIDS, Hepatitis, bacterial DNA and RNA is section and smear samples.
Solution:
A marker molecule must be designed, which will be ready and willing to serve as a key to the lock, i.e. the molecule one is looking for, much in the same way as the well known example from immune cytology, the antigen and the antibody react with each other. This marker molecule serves a chemical detector.
In the second step a molecular ballast is attached to the marker molecule to make it visible. This attachment can be an emitter of radioactivity (Radio marker) or it can radiate light (Photo marker) or it may fluoresce (Fluorescence marker=Fluorochrome). The future lies in fluorochromes.
Building this kind of detectors is not the easiest thing to do. First of all one has to know the molecule, one is searching for, in such a detail that a complementary structure, much in the key-and-lock fashion, can be designed. The key must be highly selective, i.e. it should not fit more than just the molecule searched for. Is should be able to diffuse through the interstitial spaces and within tissues and have certain life span before it gets degraded. If it supposed to act within the cell, it should be able to pass the cell membrane as well.
Such a complementary molecule, once it's been found or designed and synthesized, must in the next step get coupled with a marker ballast in such a way, that the two aspects of the result, one being the key-and-lock specificity and one the capability of making the complex visible, do not interfere with each other. And finally a process must be found, that will allow mass production of the final marker molecule.
Two lock-and-key mechanisms are at the moment biochemically available
(1) complementary DNA and RNA (cDNA, cRNA) for DNA and RNA markers
(2) antibodies when searching for proteins
What is fluorescence ?
Emission of light and heat is a natural process to distribute excess energy in a non-equilibrium systems. The process of light emission without any heat (which is rather seldom) is called luminescence. There's two modes of luminescence:
1. Phosphorescence, if a long-term emission is observed (application: storage foils)
2. Fluorescence, if the emission happens immediately after the addition of energy (=stimulation) and persists only for a short time (spontaneous emission within ca. 10-3 sec).
In case of fluorescence a short-wavelength, high-energy UV light is used to excite the electrons.
Approximately one third of the UV energy goes into the electron-pair creation. The hole in the electron cloud is immediately recombined with one of the available electrons and spontaneous emission of low-energy light with well defined spectrum (visible or infrared) occurs. It is possible to let several markers act at the same time and thus search in parallel for several substances. The resulting colors of the fluorescence characterize the markers and thus also the structures, which they dock to.
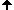 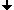 |
Short history of chromosome staining
In 1970 T. Caspersson discovered the nucleobase-specific fluorochrome quinacrin. When stained with quinacrin, each chromosome will show a specific pattern of bands, the so-called Q-bands. These bands allowed for the first time the identification of all 22 human autosomal pairs and the two human sex chromosomes. Sumner, Schnedl, Seabright discovered 1971 additional stains and preparation methods, that brought forth new banding types: G-, R-, T-, C-Bands. Dark and light bands show regions of close-packed and loose-packed DNA respectively. On top of that the banding depends on the local ratio of A-T to G-C pairs. In any case the staining bands do not have any correlation with a specific gene or group of genes. Bands have no genetic functionality, they are essentially a relatively coarse schematics of the chromosome organization.
- Caspersson,T, et al.: Differential banding of alkylating fluorochromes in human chromosomes. Exp. Cell Res. 60 (1970) 315-319
- Sumner,A., et al.: New technique for distinguishing between human chromosomes. Nature 232 (1971) 31-32
- Schnedl, W.: Analysis of the human karyotype using a reassociation technique. Chromosoma 34 (1971) 448-454
- Seabright,M.: A rapid banding technique for human chromosomes. Lancet II (1971), 971-972
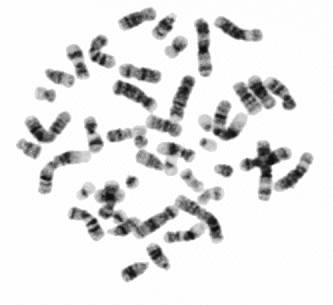
Chromosomes shown with GTG-stain
Using a known foreign DNA to hybridize and tag DNA
How did the words "fluorescence in-situ hybridization", or FISH in short, find together?
The Latin expression "in situ" describes, where the process is taking place. It means the natural, usual place, the place, where things happen, if there's no scientist around. The opposite is "in vitro", the artificial, unnatural place, like the glass container. The most natural place for chromosomes, the real in-situ place, would of course be inside a cell nucleus, which is inside a cell, which is inside a human body. Still having chromosomes on a glass slide is close enough for a geneticist to call this environment in-situ.
The Latin word "hybrida" means a bastard.. Unpaired strands of completely different origin can spontaneously hybridize, if they fit together as the key and the lock..
The basic idea of FISH is: given the unknown DNA, take a known DNA as a marker and let them hybridize. Wherever the hybridization has taken place, the unknown DNA has successfully been identified, because of its complementarity it must be identical to the known DNA.
The difficulty lies in the production of specific DNA markers. Typical DNA fragments (ca. 200 Femtograms) must be isolated from the long DNA strands. Enzymes that break the DNA chains (for instance Endonuclease DNase I, Nick-Translation) can be used for that or the chromosome can be sectioned mechanically with a fine glass needle. Then the remaining non-specific DNA must be neutralized carefully. Eventually PCR (polymerase chain reaction) is used to multiply the specific fragment (ca 1mg) and at least one fluorescence marker is added to each segment. The double-strand DNA is then separated into complementary single chains, which constitute now a sizeable amount of useful DNA marker material.
This material, either produced in-house or bought, can constitute a library of markers, that can be used to identify well-defined chromosome regions. Given such markers, the charting of unknown chromosome material proceeds as follows:
The marker and the target must first be preprocessed: the double strands must be separated, impurities cleaned out, the material denatured. The specific marker material (or possibly several of different kinds to speed up the recognition process) is added in abundance to the target to let them (given time and appropriate environment) hybridize = "in-situ hybridization". Once the process has run its course the superfluous marker material is carefully washed away to make sure there is no free, unhybridized DNA present.
What remains is a DNA hybrid, consisting half of the unknown and half of the well-known marker material. Under a fluorescence microscope the DNA tags can be brought to fluoresce and show in color where the complementary DNA has docked onto. Charting these positions on the unknown chromosome allows then comparison with existing charts.
The pioneers of the Fluorescent in-situ Hybridization of Fish for short were:
- Langer, PR., et al.: Enzymatic synthesis of Biotin-labeled polynucleotides. Proc. Natl. Acad. Sci. USA 78 (1981), 6633-6637
- Pinkel, D., et al.: Cytogenetic analysis using quantitative, high sensitivity fluorescence hybridization. Proc. Natl. Acad. Sci. USA 83 (1986), 2934-2938
6 fluorochromes in 24 markers = multiColorFISH
It is true that to discern 24 Chromosomes one needs at least 24 markers. However, 6 fluorochromes are enough, One common fluorochrome (DAPI blue counter stain) is used as usual to generally make chromosomes and their bands visible. The other 5 fluorochromes must be divided among the chromosomes in such a fashion, that each obtains its typical color. u
First publications on this subject:
- Nederlof, P., et al.: Multip. fluorescence in situ hybridization. Cytometry 11 (1990), 126-131
- Schröck, E., et al.: Multicolor spectral karyotyping of human chromosomes. Science 273 (1996), 494-497
- Speicher, M., et al.: Karyotyping human chromosomes by combinatorial multi-color FISH. Nature Genet. 12 (1996) 368-375
 |
Color table
The five columns correspond to the five indicated fluorochromes. 24 rows denote the 24 human chromosomes.
- Spectrum Orange (tm) Vysis, Inc.
- Texas Red® (tm) Molecular Probes, Inc.
- CyTM5 and Cy? Amersham Pharmacia Biotech Ltd., Inc.
6 narrow-band filters are mounted on a motorized filter wheel and used (on a fluorescence microscope, for instance Axioplan 2 Imaging mot from Carl Zeiss) to get 6 digital FL images (using for instance software ISIS/mFish or AxioVision by Carl Zeiss). The six single-color images can be viewed as one common 6Dimensional (48bit deep) color image. Given the fact that we are looking for much less (for 24 different gray levels 5 bits would suffice) an appropriate look-up table can be generated to display the eventual color-coded image of chromosomes- preferably nicely oriented and sorted out in a karyogram -.
isis/mFish (tm) MetaSystems GmbH, Altlussheim.
Axioplan 2 imaging mot (r) Carl Zeiss, Jena.
AxioVision (tm) by Carl Zeiss
|
 |
False colors representation of karyogram
Examples of arbitrary false colors:
Cy5 in Chromosom 1 is coded as white
DEAC of Chromosom 2 coded as magenta.
TexRed of Chromosom 3 coded as light green
FITC+SpeOra of Chromosome12 coded as olive green
etc...
The Karyogram shows a pathologically serious defects due to human tumor (Adenom HRT 18): the chromosomes 8, 9, 14, 17, 22 are present only single. Chromosome 19 contains parts of 10 and 3 and Chromosome Y contains the missing Chromosome 22. |
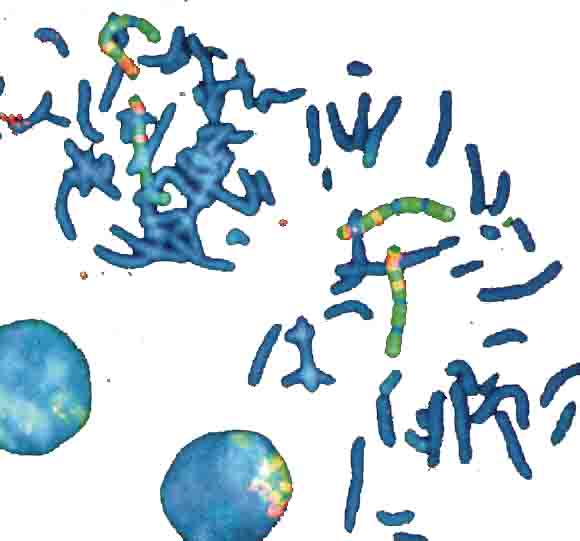
With a suitable software it is easy to determine for every pixel in the composite image, which chromosome the original point in the sample adheres to. This makes the detection of gene translocation (hopping from one chromosome to another one) possible.
High resolution multicolor banding
One can micro section single chromosomes (5 to 10 overlapping sections are needed per chromosome) and use the sections to build region-specific DNA libraries, which can be used to produce tags. These tags will be region-specific, i.e. will stain only a particular part of the subject chromosome.
First publications:
- Senger, G., et al.: Micro dissection of banded human chromosomes. Hum. Genet. 12 (1990), 507-511
- Telenius, H., et al.: Degenerate oligonucleotide-primed PCR. Genomics 13 (1992), 718-725
- Chudoba, I., et al.: High resolution multicolor banding: a new technique for refined FISH analysis of human chromosomes. Cytogenet. Cell Genet. 84 (1999), 156-160

Schematics of the PCP (Partial Chromosome Paint) staining of chromosomes 1, 5 and 7
according to I. Chudoba with 8 overlapping regional tags
Chromosome 5, PCP-stained after I. Chudoba
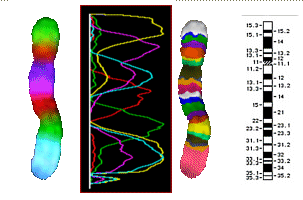
| left: |
additive color mixing of 8 tags |
| middle left |
the FL intensities of the 8 tags |
| middle right and bottom |
false colors, contrasted in a way to bring out the bands |
| right |
known chromosome 5 bands |
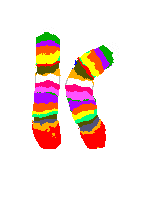
4 Chromosome sets, 2 loose in metaphase and 2 packed in nuclei
DAPI-blue-staining to show in a non-specific fasion the chromosomes.
4 chromosome pairs in a typical multi-color stain - the one in the left nucleus is barely visible, because it's covered by other chromosomes).
Picture bz V. Beensen, FSU Jena, Inst. f. Humangenetik
from Innovation Lambda4 Carl Zeiss Magazine of Microscopic Imaging and Analysis, 6/2000.
Basics of Molecular Genetics
DNA and RNA are giant molecules. DNA is visible under given circumstances in a form of tightly knotted chromosomes. The language they speak is identical for viruses, bacteriae, plants, anumals and humans. It is based on 4 letters, on four important nucleotides, which differ from each other in terms of the central base structure they posess ( Purine or Pyrimidine: Adenine/A, Thymidine/T, Guanine/G und Cytosine/C). These letters build build extremely long knotted strands of DNA, which are measured in the units of kilobases (not kilobytes). The complete length of the human genome is in the order of 3 10^9 base pairs, in other words three million kilobases.
Ca. 5 percent of the bases is used to provide the information on how to build proteins and other complex items (for instance controlling substances for the development of body parts etc). The reading of this information is again based on the key-and-lock mechanism. Because A always binds to T and C always to G, the sequences, available in DNA, will always be understood correctly: CCATCAA will always be copied into the complementary chain of GGTAGTT.
| top of page: |

